 SUR LE PLAN GENETIQUE
SUR LE PLAN GENETIQUE SUR LE PLAN CLINIQUE
SUR LE PLAN CLINIQUE SUR LE PLAN MEDICOSOCIAL
SUR LE PLAN MEDICOSOCIAL QUELQUES REFERENCES
QUELQUES REFERENCES
|
SUR LE PLAN GENETIQUE
SUR LE PLAN CLINIQUE
SUR LE PLAN MEDICOSOCIAL
QUELQUES REFERENCES
|
|
||
|
|
|
|
|
|
|
|
 NOTIONS DE BASE
NOTIONS DE BASEtexte extrait du compte-rendu de nos journées d'information sur les anomalies chromosomiques rares Le caryotype est l'examen des chromosomes qui permet d'étudier leur nombre et leur structure. L'être humain possède au sein de chaque cellule (sauf dans les ovules et les spermatozoïdes), 23 paires de chromosomes, soit 46 au total qui se répartissent :
On utilise une nomenclature précise pour désigner les anomalies
chromosomiques.
Quand on regarde le chromosome au microscope, celui-ci ressemble à un grand X, constitué :
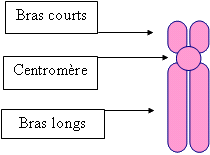
schéma conçu par Thierry Marchetti pour les comptes-rendus de nos journées d'informations Pour les repérer précisément sur le chromosome, des chiffres sont notés après la lettre " p " ou " q " :
 schéma conçu par Thierry Marchetti pour les comptes-rendus de nos journées d'informations
Un tableau clinique dû à cette délétion a été décrit en 1963 pour la 1ère fois par le généticien français M. Lejeune et ses collaborateurs. Il remarqua qu'un certain nombre d'enfants qu'il avait vu, pleuraient en poussant des cris aigus très particuliers comme le miaulement d'un chat. Ce cri constitue un critère important de diagnostic et il disparaît vers l'âge de 3 ans bien que la voix conserve un registre aigu. Le syndrome du Cri du Chat est une anomalie chromosomique de structure. Une petite partie du matériel génétique des bras courts du chromosome 5 est absente : elle a été délétée. Ainsi, on parle également de " délétion 5p " ou " 5p- ", ou encore de " monosomie partielle " du chromosome 5p car seul un fragment chromosomique manque et non pas sa totalité. Pour que le syndrome du cri du chat apparaisse, une zone dite critique doit être contenue dans la délétion, c'est la région 5p15.2 (région 1 bande 5 sous bande 2 des bras courts du chromosome 5). La délétion peut être :
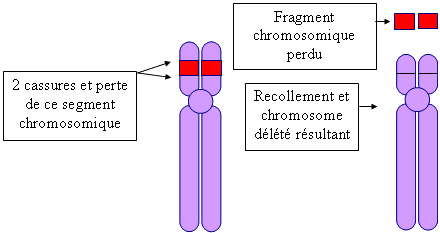 schéma conçu par Thierry Marchetti pour les comptes-rendus de nos journées.
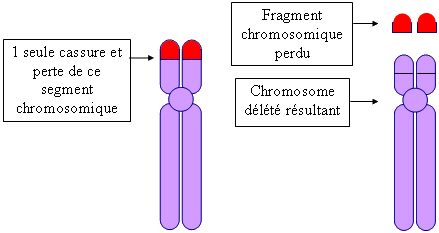 schéma conçu par Thierry Marchetti pour les comptes-rendus de nos journées.  FREQUENCE
FREQUENCESelon les bases de données, le taux de prévalence est estimé entre 1/15.00 et 1/50.000.  DIAGNOSTIC
DIAGNOSTICQuand un médecin soupçonne le syndrome du cri du chat chez un bébé, il est nécessaire de confirmer son diagnostic par un examen chromosomique, le caryotype. image fournie par les généticiens lors de nos journées d'informations Dans la grande majorité des cas (85 %), il s'agit d'un problème génétique survenu de manière " accidentelle " ou dite " de novo " chez l'enfant. Si tel est le cas, le risque de récidive en cas de future(s) grossesse(s) est a priori très faible. Toutefois, pour diverses raisons, on peut conseiller aux parents de pratiquer un examen prénatal. Moins souvent (15 %), l'anomalie chromosomique de l'enfant est héritée, en raison de l'existence d'un remaniement chromosomique équilibré (sans perte ni gain de matériel génétique) chez l'un des parents : translocation, insertion, inversion qui inclue la région 5p15.2. Dans ce cas, il existe un risque potentiel de récurrence qu'un généticien doit évaluer au cas par cas et l'examen prénatal sera proposé systématiquement à la mère.  CONSEIL GENETIQUE
CONSEIL GENETIQUEMême si le diagnostic a été annoncé par un pédiatre ou un neurologue, il est important que ce résultat génétique soit expliqué par le spécialiste de la génétique, c'est-à-dire un généticien :
|
||
|
|
||
 DEVELOPPEMENT
DEVELOPPEMENT
|
 PRISE EN CHARGE
PRISE EN CHARGEMEDICALE |
 PRISE EN CHARGE
PRISE EN CHARGE MEDICOSOCIALE |
|
LE DEVELOPPEMENT Les parents constatent que les bébés ayant le cri du chat, ont à la naissance un poids, une taille et un périmètre crânien en général plus petits que les autres enfants. Le cri et les pleurs du bébé sont caractéristiques. Les bébés ont souvent des difficultés pour s'alimenter. Ils tètent mal, boivent lentement, font des fausses-routes et ils régurgitent beaucoup. Les parents notent également des problèmes respiratoires, de vision tels que cataracte, myopie, strabisme mais aussi de temps en temps des malformations cardiaques, rénales et intestinales congénitales. Certaines de ces malformations pourront être résolues par une intervention chirurgicale. Il est donc nécessaire que tous ces enfants soient bien examinés sur ces points. L'hypotonie (diminution du tonus musculaire) est souvent présente dès le début avec apparition, au cours du développement, d'une hypertonie des membres (augmentation pathologique du tonus musculaire), de réflexes vifs et d'une démarche spastique. Par la suite, les enfants apprennent à jouer et à manger seul. Toutefois, ils mangent des repas moulinés pendant parfois plusieurs années car ils ne savent pas mâcher. Mais les parents disent qu'avec l'âge, cela semble s'améliorer. La constipation est souvent évoquée, tout comme les troubles du sommeil. Leur développement moteur est plus lent que celui des autres enfants du même âge. Lever la tête, se retourner, attraper les objets, apprendre à s'asseoir et ramper, se tenir debout et marcher demande plus d'efforts et de temps pour l'acquérir. Ainsi, les parents remarquent que la plupart des enfants ayant ce syndrome apprennent à s'asseoir, ramper, se tenir debout et marcher. Quelques-uns ont une motricité tellement limitée qu'ils doivent se déplacer en chaise roulante. Une scoliose peut-être l'une des origines de ce problème. Ce sont des enfants dont le caractère est joyeux, qui sont attachants et qui peuvent être très têtus. Ils sont souvent vifs, de bonne humeur, curieux, intéressés par leur entourage et ils se lient facilement avec d'autres personnes. Le contact social et la communication sont bons, même si certains enfants ne parlent pas. Au niveau des loisirs, les parents constatent qu'ils aiment l'eau, les promenades, la musique et le chant, le poney, les histoires, la danse, ... PRISE EN CHARGE MEDICALE La 1ère année est souvent difficile pour les parents entre le diagnostic, les problèmes de l'enfant et la prise en charge médicosociale qu'il faut organiser : kinésithérapeute, psychomotricien, ergothérapeute, orthophoniste sont les rééducations de base. D'autres spécialités médicales seront ajoutées en fonction des pathologies de l'enfant : neuropédiatre, ORL, ophtalmologue, psychologue, cardiologue, gastroentérologue, orthopédiste, ... Au niveau de la communication, les parents disent que souvent les 1ers mots n'apparaissent pas avant 4 ou 5 ans. Comme certains enfants ne parlent pas du tout, la communication à l'aide de gestes et d'images peut aider l'enfant à se faire comprendre. L'aide d'un logopède dans cette phase du développement est presque toujours nécessaire. Certains appareillages spécifiques pourront aider l'enfant à faire certaines acquisitions ou bien pour son confort quotidien. PRISE EN CHARGE MEDICO-SOCIALE
Selon les pays, diverses aides financières, médicales ou sociales existent. Il est donc important que les parents se renseignent sur leur existence. En France, notons les principales comme :
|
||
|
|
Site francophone sur le syndrome du cri du chat |
Dernière modification le : 2011 |
Fermer la fenêtre |


